
Genome Maintenance and Quantitative Proteomics
Cells use proteins to do many tasks, including the replication and segregation of their genome during each cell division. No single protein is capable of these tasks alone - dozens of proteins must assemble into distinct protein complexes that undergo dynamic turnovers. Our work falls into three areas. First, the regulatory mechanisms that control DNA replication and DNA damage response. Second, the controls of kinetochore assembly and disassembly during the cell cycle. Third, the development of Concatemer-Assisted Stoichiometry Analysis (CASA) for quantitative analysis of protein complexes and their modifications.
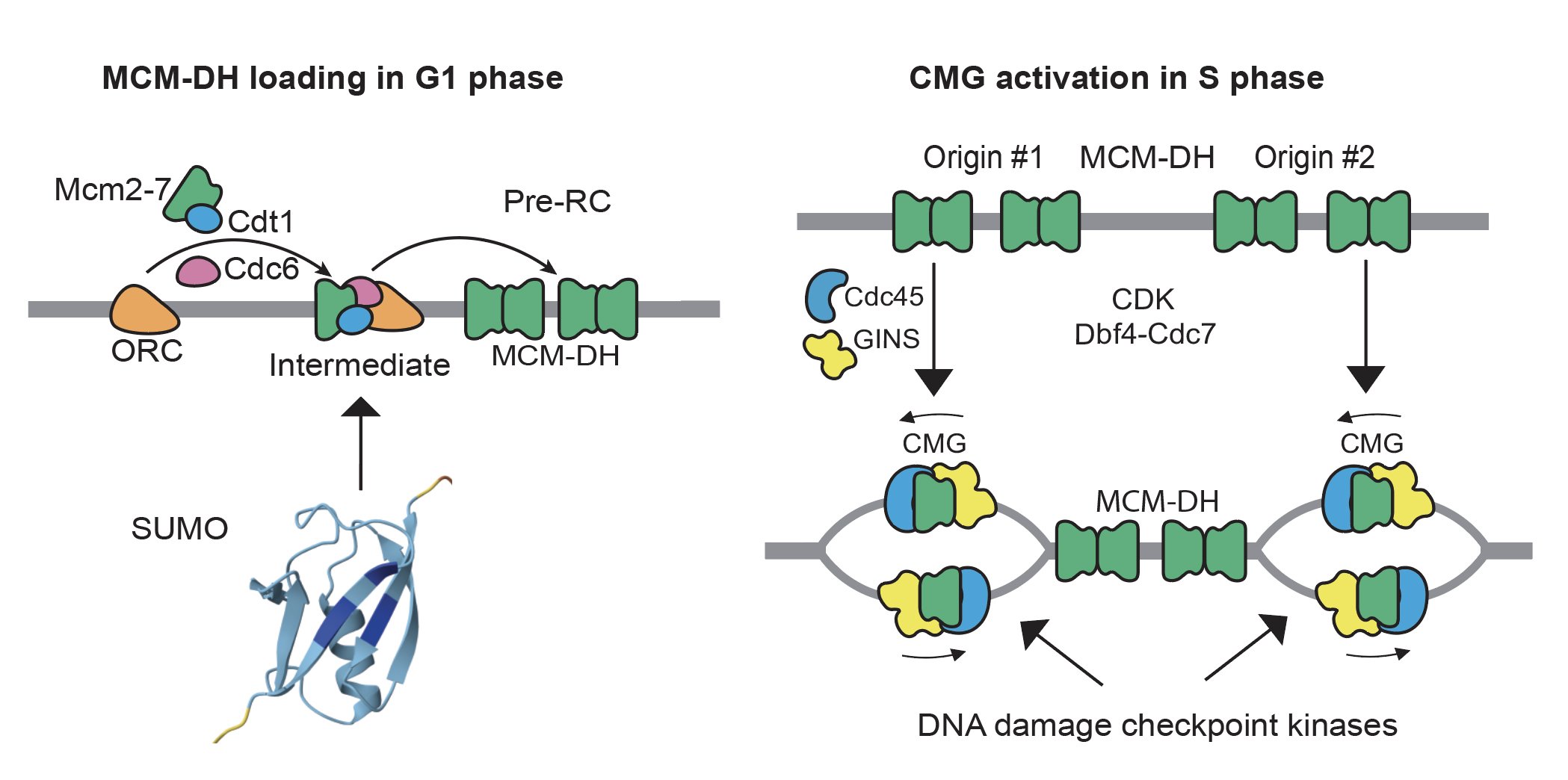
Although how individual MCM is loaded and activated to form the CMG helicase have been extensively studied and well understood, their cellular controls are not. We have a long-standing interest in the role of DNA damage checkpoint kinases in preventing genome rearrangements through the control of DNA replication, which remains incompletely understood. We have also investigated the role of SUMO, a small ubiquitin-like protein, in preventing genome rearrangements (Albuquerque 2013 and 2016, Liang 2018, Suhandynata 2021, and Quan 2022). More recently, we focus on how DNA replication factors are assembled on chromatin during chromosome replication (Wang 2024), and how this is controlled by various post-translational modifications.
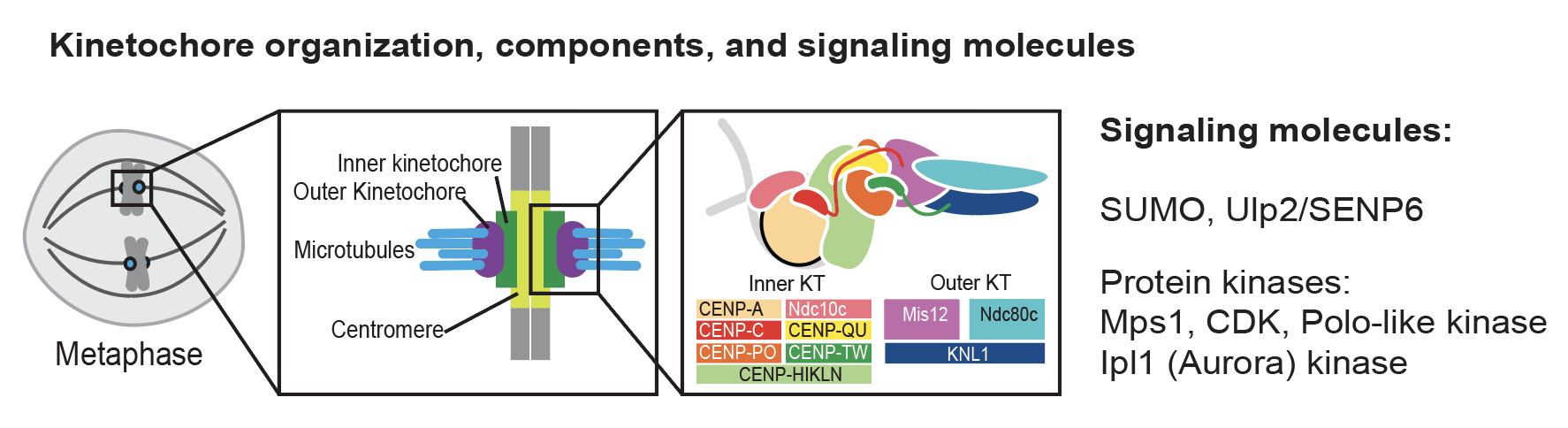
Kinetochores control chromosome segregation by connecting chromosomal centromeres to the spindle microtubules and scaffolding many signaling enzymes to prevent mis-segregation. Despite extensive studies, we still do not have a complete understanding of how kinetochores are assembled and how this is controlled. Our recent studies established a direct connection between the Ulp2 desumoylase and the inner kinetochore (Suhandynata 2019, Quan 2021), a critical role of protein phosphorylation in controlled Mif2-dependent kinetochore assembly (Hinshaw 2023), and an essential connection within the inner kinetochore (Deng 2023). We continue to study how kinetochores are assembled on native centromeres during the cell cycle and how various post-translational modifications control this fundamental process.
Concatemer Assisted Stoichiometry Analysis (CASA)
For the past three decades, mass spectrometry (MS) has seen numerous biological applications. Improvements of scan speed, sensitivity and mass resolution have stimulated a wide adoption of discovery-based MS. However, there is a hidden cost: discovery-based MS favors the detection of more abundant proteins, while lesser abundant proteins are subjected to greater uncertainty in precision and accuracy, including interference from more abundant proteins. This deficiency is particularly dire since most cell biological processes involve dynamic turnovers of lower abundance proteins. Discovery-based MS is ill prepared to acquire such knowledge.
The development of CASA addresses this challenge via targeted MS (Cai, 2024). Unlike discovery-based MS, targeted MS measures peptides via their unique “fingerprints” - the fragment ions that are produced by MS. To establish such fingerprints, CASA generates isotope-coded peptide standards, each serving as “GPS” to guide their detection. The outcome is dramatic – CASA, equipped with targeted MS, achieves unprecedented accuracy, sensitivity and precision in measuring low abundant proteins that are prevalent in many cell biological processes.